Chapter 11: Your Body's Obstacle Course
Chapter 11: Your Body’s Obstacle Course
Part IV: What Your Body Does With Coffee
A few months ago, I was having dinner with my friend Laura — a journalist, decidedly not a scientist — and she asked me what I was working on. I told her I was studying how coffee compounds behave inside the human body. She looked at me with the kind of polite blankness that tells you someone is already formulating a way to change the subject. So I tried a different approach.
“You know how when you take a pill,” I said, “only a fraction of it actually reaches the part of your body where it needs to work?”
She nodded. She’d had this experience — taking ibuprofen and waiting for it to kick in, wondering where it was and what was taking so long.
“Your body is basically an obstacle course for molecules,” I continued. “Every compound you swallow has to survive your stomach acid, cross the wall of your intestine, travel through your blood without getting destroyed by your liver, reach wherever it needs to go, and then — ideally — not poison you in the process. Most molecules fail somewhere along the way.”
Laura put down her fork. “And the stuff in coffee goes through this?”
“Every single compound. Every time you take a sip.”
That conversation is really the heart of this chapter. Because when we talk about what coffee “does” to you — whether it wakes you up, protects your liver, affects your heart — we are implicitly assuming that the molecules responsible can actually survive the obstacle course. That they can get from your cup to their target. And that is not a trivial assumption. It is, in fact, the central question that the pharmaceutical industry spends billions of dollars trying to answer for every drug candidate it develops.
In this chapter, I want to take you inside that obstacle course. I want to show you the framework that pharmaceutical scientists use to evaluate whether a molecule has any chance of working in the human body, and then I want to apply it — carefully, honestly, with full acknowledgment of its limitations — to the compounds in your coffee.
The Five Checkpoints: Introducing ADMET
The pharmaceutical industry has a name for the obstacle course. They call it ADMET, and it stands for five stages that every molecule must navigate:
Absorption. Can the molecule get through your gut wall and into your bloodstream? Picture the lining of your small intestine: a single layer of cells, each wrapped in a fatty membrane, packed shoulder-to-shoulder like bouncers at a velvet rope. A molecule that is too large, too electrically charged, or too water-loving gets turned away. It continues down the gut and exits the way it came in — straight through you, never absorbed, no matter how powerful it looked in a test tube.
Distribution. Once a molecule slips into the blood, can it navigate to the organ where it needs to act? Your bloodstream is not an open highway — it is more like a city with checkpoints. Albumin and other blood proteins grab passing molecules and hold them like toll collectors. Specialized barriers guard critical organs: the blood-brain barrier, the placental barrier, the blood-testis barrier. A molecule might be beautifully absorbed and still spend its entire life stuck in transit, circling in the blood, never reaching the tissue that matters.
Metabolism. Your liver is a demolition crew that never clocks out. The moment a foreign molecule arrives via the portal vein — and the portal vein is the first stop after gut absorption, meaning the liver gets first crack at everything you swallow — enzymes begin tearing it apart. This is enormously useful when the foreign molecule is a toxin. It is less useful when it is a compound you want to deliver somewhere else. Metabolism is the clock ticking down: if the liver works too fast, the molecule is rubble before it can do anything meaningful.
Excretion. Your kidneys, your liver, your lungs, even your skin — your body has multiple exits, and every molecule is being pushed toward one of them from the moment it enters. Think of it as a stadium that empties continuously: the ushers never stop working. If a compound is cleared too quickly, its concentration in the blood never builds up enough to have an effect. It is gone before it arrives.
Toxicity. And finally, the security check. Is the molecule safe? Does it damage cell membranes, nick DNA strands, accumulate in your kidneys or liver until it poisons the organ that was supposed to process it? A compound can pass the first four stages with flying colors and still fail catastrophically at the fifth. The pharmaceutical graveyard is full of molecules that were brilliantly absorbed, perfectly distributed, and lethally toxic.
I like to think of ADMET as airport security for molecules. Absorption is getting through the entrance — you need the right ticket, the right size of luggage. Distribution is navigating the terminal to find your gate — there are barriers, wrong turns, places you are not allowed to go. Metabolism is the clock on your boarding pass — you have a limited window before your flight leaves without you. Excretion is the exit on the other side — once you are through, you are gone. And toxicity is the security screening itself — you can have a perfectly valid ticket and still get pulled aside if you are carrying something dangerous.
Every drug that has ever reached your pharmacy shelf has passed all five stages. And every drug candidate that failed — and the failure rate in pharmaceutical development is staggering, well above ninety percent — stumbled at one or more of these checkpoints.
Figure 11.1. Computational ADMET heatmap: simulation-based inference reveals how coffee's bioactive compounds navigate the body's obstacle course, with predicted scores for absorption, distribution, metabolism, and excretion.
Now. What does this have to do with coffee?
Everything, as it turns out. Because the compounds in coffee are not drugs. Nobody designed them. Nobody optimized them for human absorption. They evolved in the coffee plant for entirely different reasons — mostly to deter insects, attract pollinators, or protect against ultraviolet radiation. And yet, when we subject them to the same ADMET screening that pharmaceutical companies use for drug candidates, something rather remarkable emerges.
But before we get to coffee specifically, I need to tell you about a man named Christopher Lipinski and the simple set of rules that transformed drug development.
The Molecular Passport: Lipinski’s Rule of Five
In the late 1990s, Christopher Lipinski was a medicinal chemist at Pfizer, one of the world’s largest pharmaceutical companies. And Pfizer, like every other drug company at the time, had a problem. They were spending enormous sums of money developing compounds that looked spectacular in the laboratory — they bound to their targets beautifully, they killed cancer cells in petri dishes, they inhibited enzymes with exquisite precision — and then they failed, over and over, when they were actually given to patients.
The compounds simply were not getting absorbed. They were failing at the very first checkpoint of the obstacle course. And nobody had a quick way to predict, early in development, which molecules would be absorbed and which would not.
Before Lipinski published his rules, the pharmaceutical industry was flying somewhat blind. Drug companies would identify a promising compound, spend years and enormous sums optimizing its potency against a biological target, push it into animal studies and then human trials — only to discover that the molecule could not be absorbed from the gut in sufficient quantities to work. The compound was potent in a test tube and useless in a patient.
This problem was not occasional. It was endemic. Estimates vary, but poor pharmacokinetics — the science of how drugs move through the body — was responsible for a substantial proportion of all drug development failures. Each failure could represent years of work and hundreds of millions of dollars.
Lipinski’s contribution was not a new technology or a new class of drugs. It was something simpler and, in its way, more powerful: a filter. A set of rules that could be applied at the very beginning of the development process — before the expensive studies, before the clinical trials — to identify molecules that were unlikely to be absorbed. By screening out poor candidates early, the industry could focus its resources on compounds that had a realistic chance of working in the human body.
The impact was transformative. Lipinski’s Rule of Five became one of the most cited papers in pharmaceutical science, and some version of his filters is now applied in virtually every drug development program in the world. It did not solve the problem of drug failure — many other things can go wrong — but it addressed one of the most wasteful and preventable sources of failure in the industry.
Lipinski did something that seems, in retrospect, almost embarrassingly straightforward. He gathered data on thousands of drugs that had successfully made it through clinical trials and into pharmacies. Then he asked: what do these successful molecules have in common?
The answer was four properties, all of which happened to cluster around the number five — which is why his discovery became known as the Rule of Five. A molecule is more likely to be absorbed orally if it meets these criteria:
Molecular weight no greater than 500 daltons. A dalton is the unit used to measure molecular mass. Molecules heavier than 500 daltons tend to be too large to cross the intestinal wall efficiently. Think of it as a size limit on your luggage — too big, and it does not fit through the door.
LogP no greater than 5. LogP is a measure of how much a molecule prefers oil over water. (The “log” means it is on a logarithmic scale, so small changes in the number represent large changes in the property.) Molecules need some lipophilicity — some oil-loving character — to cross the fatty membranes of your cells. But too much lipophilicity, and they get stuck in the membranes instead of passing through. A LogP of 5 or less hits the sweet spot.
No more than 5 hydrogen bond donors. Hydrogen bonds are the sticky interactions that molecules form with water. Donors are the parts of a molecule that can offer a hydrogen atom to form such a bond. Too many donors make a molecule too “sticky” with water, which prevents it from crossing the oily cell membranes of the gut.
No more than 10 hydrogen bond acceptors. Acceptors are the other side of hydrogen bonding — the parts that can receive a hydrogen atom. The same principle applies: too many acceptors, too much water stickiness, too little membrane crossing.
That is it. Four numbers. Molecular weight at or below 500. LogP at or below 5. Hydrogen bond donors at or below 5. Hydrogen bond acceptors at or below 10. A molecule that meets all four criteria holds what I like to call a “molecular passport” — a set of credentials that says, in effect, this molecule has the physical and chemical properties needed to cross biological membranes and be absorbed from the human gut.
It is elegant in its simplicity. It is also, and I want to be very clear about this, a screening tool, not a guarantee. Many molecules that pass Lipinski’s Rule still fail as drugs for other reasons. And some successful drugs break one or more of the rules. Lipinski himself was always careful to present this as a guideline, not a law. But as a first filter — as a way to quickly evaluate whether a molecule has the basic properties needed for oral bioavailability — it remains one of the most useful tools in pharmaceutical science.
Applying the Passport Check to Coffee
Now we arrive at the question that motivated this chapter. If Lipinski’s Rule of Five is the pharmaceutical industry’s first filter for drug candidates, what happens when we apply it to the bioactive compounds in coffee?
I should say that when I first ran this analysis, I was not sure what to expect. Coffee compounds are not drugs. They were not designed to be absorbed by humans. There was no particular reason to assume they would pass a filter that was developed for pharmaceutical optimization.
And yet all fifteen of the key bioactive compounds in coffee pass Lipinski’s Rule.
Every single one. Fifteen for fifteen. A clean sweep from a plant that never once consulted a medicinal chemist. I actually laughed — the kind of laugh that escapes when something defies your expectations so completely that surprise bypasses everything else on its way out.
I remember staring at the spreadsheet when the numbers came back, scrolling down the column of pass/fail flags, waiting for the first failure. It never came. Outside my window, someone was grinding espresso in the café across the street — I could hear the burr whirring through the open glass — and I remember thinking: you have no idea what you are. Let me walk you through a few examples to give you a sense of why that result stopped me in my tracks.
Caffeine — the compound everyone knows — has a molecular weight of just 194 daltons. That is well under the 500-dalton ceiling, making it a small molecule by pharmaceutical standards. It is also moderately lipophilic, meaning it has just the right balance of oil-loving and water-loving character to cross cell membranes efficiently. If caffeine were a drug candidate arriving at Lipinski’s filter, it would sail through without a second glance.
Cafestol and kahweol — the two diterpene compounds found primarily in unfiltered coffee — are larger, with molecular weights of 316.4 and 314.4 daltons respectively. These are still comfortably within the Lipinski window, though they sit in the middle range rather than the low end. Their lipophilic character, which makes sense given that they are found in coffee’s oily fraction, also falls within acceptable limits.
Chlorogenic acids, the large family of polyphenols that give coffee its astringent bite, also pass the molecular weight and hydrogen bonding criteria, though they push closer to some of the boundaries. We will come back to chlorogenic acids in the next chapter, because their story gets more complicated when we look at distribution — specifically, whether they can reach the brain.
The other compounds — trigonelline, ferulic acid, caffeic acid, quinic acid, and the rest of the fifteen — all fall within the Lipinski parameters as well. It is a clean sweep.
Caffeine's ADMET Profile — Why It Works So Well
Absorption: Near-complete oral bioavailability (~99%) — almost everything you drink reaches your bloodstream.
Distribution: Crosses the blood-brain barrier easily (low molecular weight, moderate lipophilicity).
Metabolism: Processed by CYP1A2 in the liver — your genetics determine if you're a fast or slow metabolizer.
Excretion: Half-life of 3-7 hours (varies by CYP1A2 genotype, pregnancy, smoking status).
Toxicity: LD₅₀ estimated at ~150-200 mg/kg — you'd need ~75 cups in rapid succession. Your morning three are safe.
Why This Is Surprising (and Why It Is Not)
There are two ways to react to this result.
The first reaction, the one that tempts me as a scientist who finds coffee endlessly fascinating, is to be impressed. Here are fifteen compounds, produced by a plant for its own purposes, and every one of them happens to have the molecular properties that pharmaceutical chemists spend years trying to engineer into drug candidates. That is not nothing.
The second reaction, the more measured one — and the one I arrived at roughly forty-eight hours later, after the initial giddiness wore off — is to note that this is less surprising than it first appears. Coffee compounds are, for the most part, small molecules. They are the products of well-characterized biochemical pathways — the phenylpropanoid pathway for chlorogenic acids, the purine pathway for caffeine, the terpenoid pathway for cafestol and kahweol. These pathways tend to produce molecules in the low hundreds of daltons, with moderate lipophilicity and limited hydrogen bonding. In other words, they tend to produce molecules that fall naturally within the Lipinski window, not because they were selected to be bioavailable in humans, but because the underlying chemistry of these pathways generates molecules of that general size and character.
It is also worth noting what this result does not apply to. Coffee contains many compounds beyond the fifteen small-molecule bioactives we have been focusing on. Most notably, it contains melanoidins — the large, brown, complex polymers formed during roasting through the Maillard reaction. And melanoidins do not come anywhere close to passing Lipinski’s Rule.
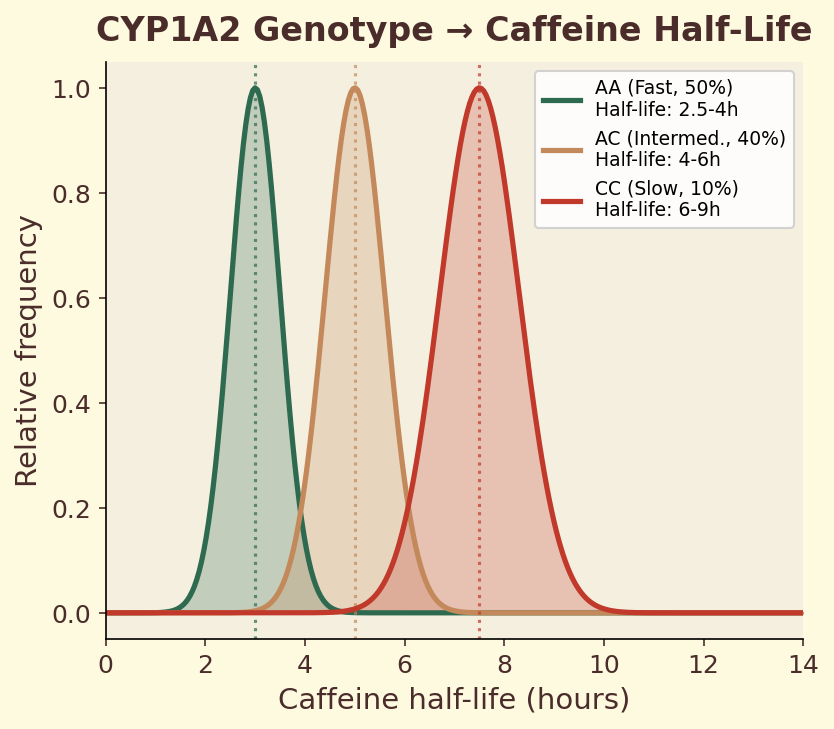
Figure 11.2a. Caffeine half-life distributions: population-level variation in caffeine elimination half-life by CYP1A2 genotype, from 2-3 hours (fast) to 6-9 hours (slow).
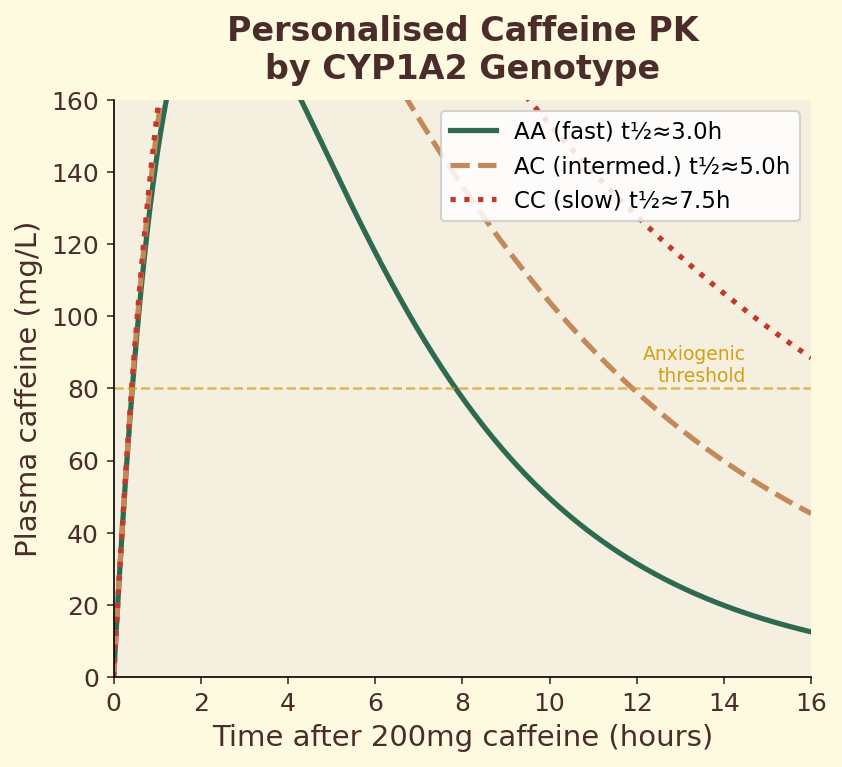
Figure 11.2b. Personalised pharmacokinetics: simulated blood caffeine curves for AA, AC, and CC genotypes over 24 hours following morning coffee consumption.
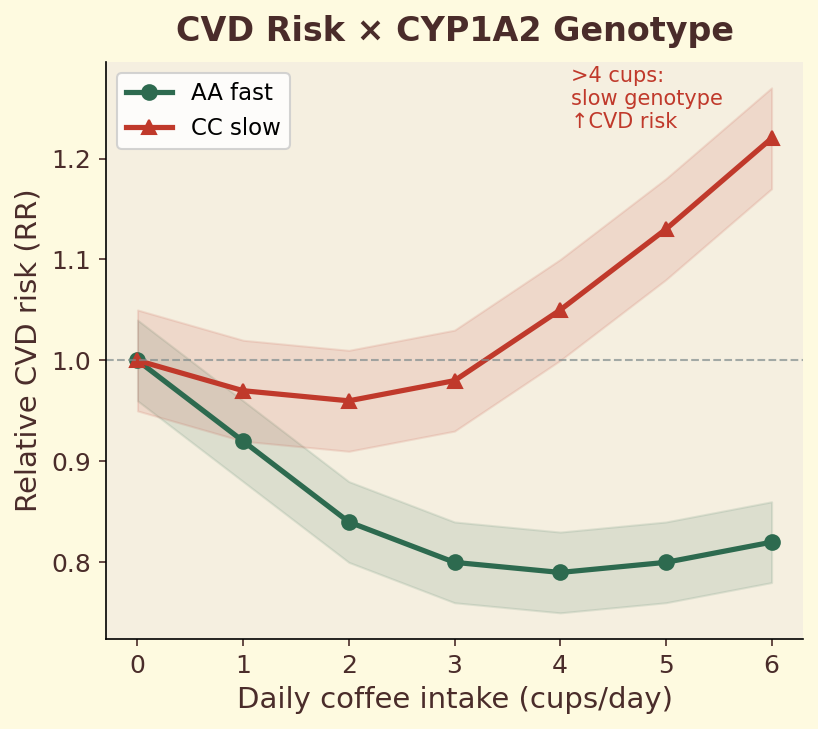
Figure 11.2c. Cardiovascular risk: interaction between CYP1A2 genotype and daily coffee intake on CVD risk, showing divergent dose-response curves for fast versus slow metabolizers.
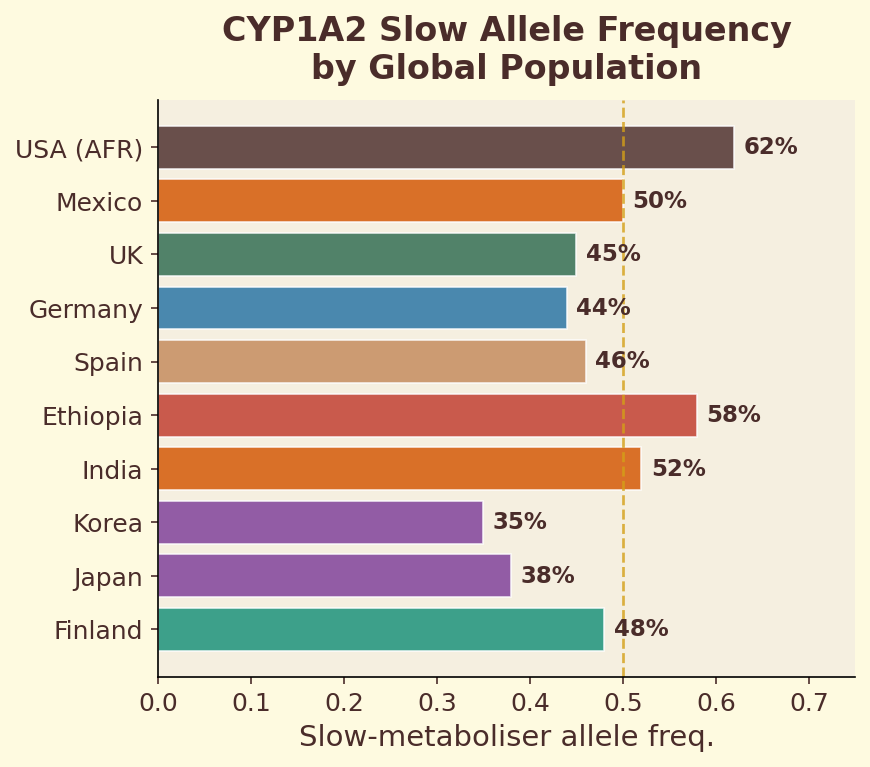
Figure 11.2d. Global allele frequency: geographic distribution of the CYP1A2 slow-metabolizer allele across world populations.

Figure 11.2e. Key SNPs: summary table of genetic variants relevant to personalised coffee nutrigenomics, including CYP1A2, ADORA2A, and AHR polymorphisms.
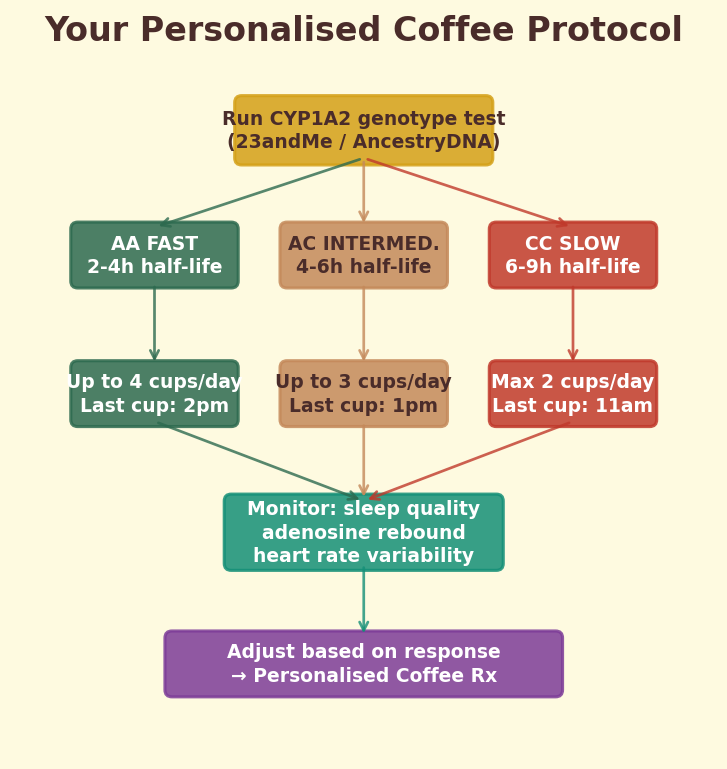
Figure 11.2f. Personalised protocol: decision flowchart for optimising coffee intake based on genotype, sleep sensitivity, and cardiovascular risk factors.
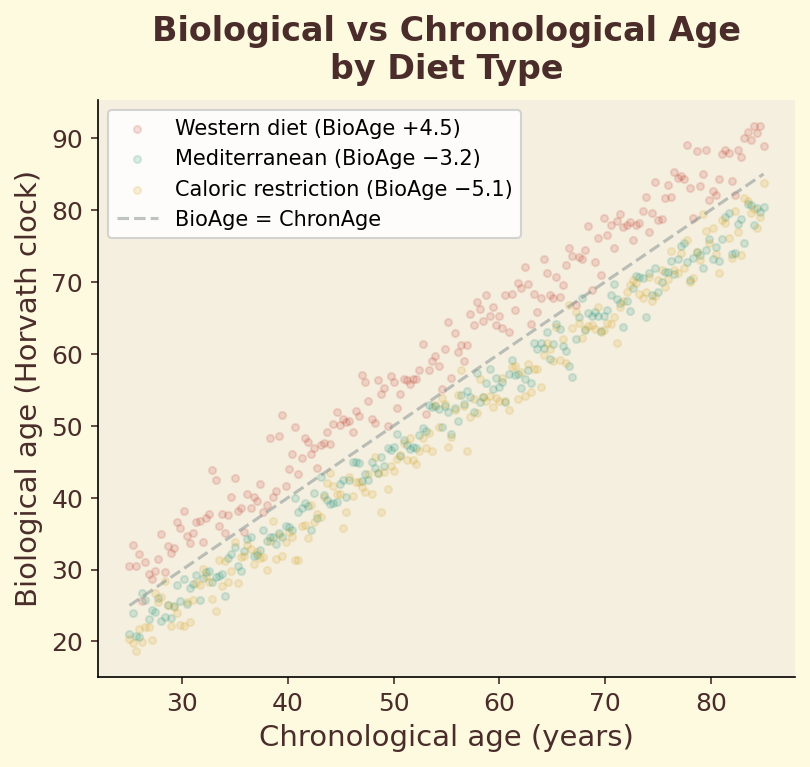
Figure 11.3a. Biological versus chronological age: scatter plot showing how different dietary patterns shift the relationship between measured epigenetic age and calendar age.
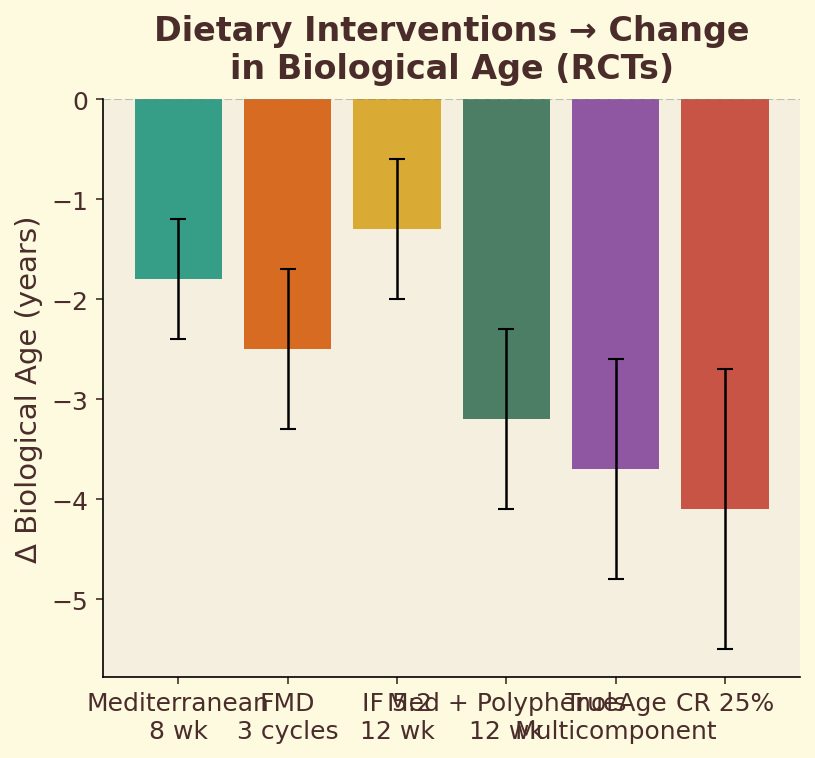
Figure 11.3b. Dietary interventions: ranked effect sizes of major dietary patterns on biological age deceleration, from Mediterranean to Western diets.
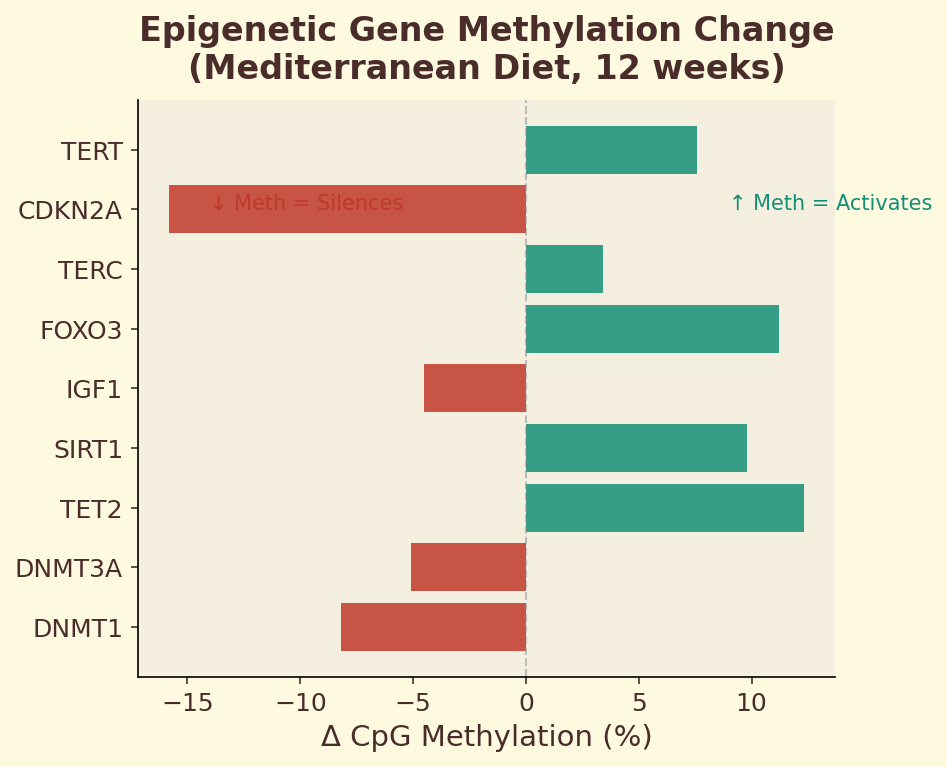
Figure 11.3c. CpG methylation changes: specific genomic loci showing altered methylation status after 12 months of Mediterranean diet adherence, highlighting inflammation and antioxidant defense genes.
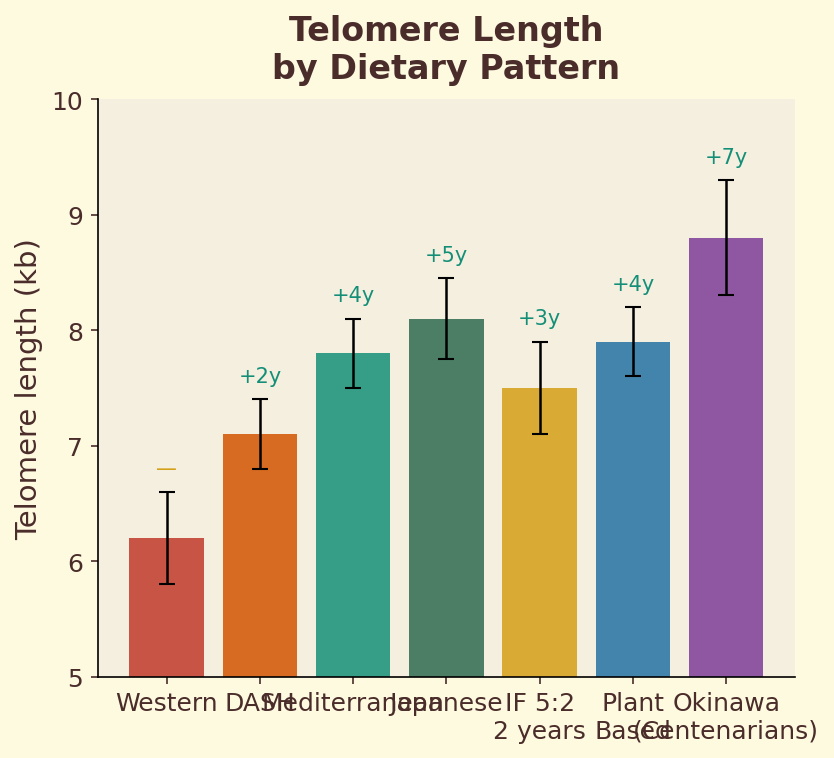
Figure 11.3d. Telomere length: relative leukocyte telomere length across dietary pattern quintiles, showing longer telomeres in populations with higher polyphenol and antioxidant intake.
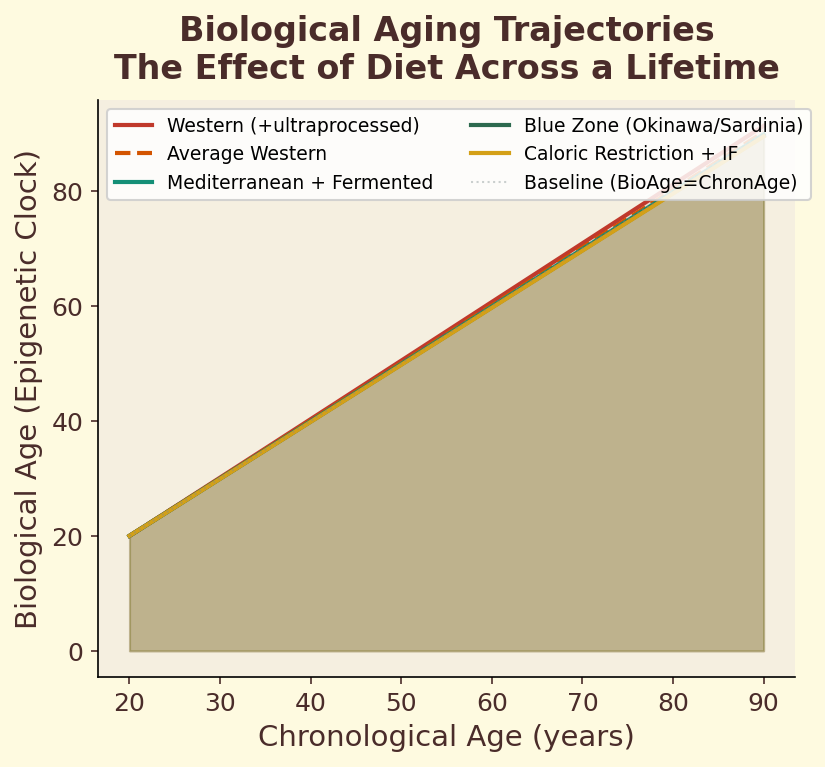
Figure 11.3e. Aging trajectories: projected biological aging curves across four decades for different dietary patterns, illustrating cumulative divergence between accelerated and decelerated aging.
Melanoidins are the most abundant compounds in brewed coffee by weight. They are responsible for much of coffee’s brown color, its body, and its capacity to act as dietary fiber in the gut. They are also, from an ADMET perspective, essentially invisible.
The reason is simple: melanoidins are enormous. Their molecular weights range from roughly 3,000 daltons for the smallest fragments to well over 100,000 daltons for the larger polymers. Lipinski’s cutoff is 500 daltons. Melanoidins exceed it by an order of magnitude at minimum and by several orders of magnitude at maximum.
This means melanoidins are not absorbed intact through the gut wall. They do not enter the bloodstream as whole molecules. They do not distribute to organs. In pharmaceutical terms, they are not orally bioavailable — at least not in their intact polymeric form.
This does not mean melanoidins are biologically inert. They interact extensively with the gut microbiome, they may be partially broken down into smaller absorbable fragments, and they have well-documented effects on digestive health. But those effects happen in the gut, not in the bloodstream or the brain. When we talk about ADMET screening and coffee’s bioactive compounds, we are talking exclusively about the small-molecule fraction — the caffeine, the chlorogenic acids, the diterpenes, the phenolic acids. The melanoidins are playing a different game entirely.
I mention this because it is important to be precise about what ADMET screening can and cannot tell us. It is a framework for evaluating small molecules that are intended to be absorbed and distributed through the body. Applying it to melanoidins would be meaningless — like trying to board an airplane with a shipping container.
The Pharmaceutical Lens
Let me be explicit about what we are doing in this chapter, because the framing matters.
We are taking a tool developed by and for the pharmaceutical industry — ADMET screening, beginning with Lipinski’s Rule of Five — and applying it to coffee compounds. We are asking: If these fifteen bioactives were drug candidates, would they pass the first round of screening?
The answer is yes. All fifteen have molecular properties — size, lipophilicity, hydrogen bonding capacity — that are consistent with oral bioavailability. In pharmaceutical terms, they look like plausible drug candidates. They hold valid molecular passports.
This is a meaningful finding. It tells us that the physical and chemical properties of these molecules are compatible with absorption from the gut. It tells us that there is no obvious structural barrier — no excessive size, no extreme water solubility, no impossible charge distribution — that would prevent them from entering the bloodstream.
But I want to be equally explicit about what this finding does not tell us.
What ADMET Screening Does Not Tell Us
Passing Lipinski’s Rule is necessary but not sufficient for a compound to have biological effects. It is the first filter, not the last. And the distance between “passes the first filter” and “has proven clinical effects in humans” is vast. Let me enumerate what our analysis has not established.
It does not tell us how much is absorbed. Lipinski’s Rule predicts that absorption is possible, not that it is complete. The actual fraction of a compound that crosses the gut wall depends on many factors beyond molecular properties — the formulation, the food matrix, the individual’s gut health, the presence of efflux pumps that actively push molecules back out, and much more. A molecule can pass Lipinski’s Rule and still have very low actual absorption.
It does not tell us about concentrations. Even if a coffee compound is absorbed, the amount that reaches any particular organ depends on the dose (how much was in the cup), the absorption efficiency, the rate of metabolism, and the rate of excretion. These are pharmacokinetic questions that cannot be answered by molecular property analysis alone. They require actual measurement in actual humans — the kind of clinical pharmacokinetic studies that have been done thoroughly for caffeine but much less thoroughly for most other coffee compounds.
It does not tell us about biological activity. A molecule that reaches its target is not necessarily a molecule that does anything at that target. Biological activity requires the molecule to interact with specific proteins, receptors, enzymes, or other biological structures in a way that changes their function. ADMET screening says nothing about whether such interactions occur.
Still with me? One more gap — and this one is the widest of them all.
It does not tell us about dose-response. Even if a compound is absorbed, reaches its target, and has biological activity, the question remains: at what concentration? The concentrations achieved by drinking a cup of coffee may be far below the threshold needed for meaningful biological effects. Or they may be right in the sweet spot. Without dose-response data from human studies, we simply do not know for most compounds.
I want to be very clear about this, because it matters for everything that follows in this book: showing that coffee compounds have drug-like molecular properties is not the same as showing they act as drugs. Lipinski’s Rule tells us that the door is open. It does not tell us what is on the other side.
I have seen too many popular science articles that leap from “this compound has drug-like properties” to “this compound will cure your disease.” That leap skips over approximately fifteen years of clinical development, billions of dollars in research costs, and a failure rate that would make any gambler weep. I am not going to make that leap. What I am going to do is follow the evidence through each stage of the obstacle course, reporting what we know, what we suspect, and what we simply do not yet understand.
A Framework, Not a Verdict
Perhaps the most important thing to take away from this chapter is the framework itself. ADMET thinking — the habit of asking, for every compound, Can it be absorbed? Can it reach its target? Is it metabolized? Is it excreted? Is it safe? — is a discipline. It forces you to think systematically about what happens between the moment a molecule enters your mouth and the moment it (potentially) has an effect.
Most popular writing about coffee skips this entirely. You read that “coffee contains antioxidants” as if the mere presence of antioxidants in the cup guarantees antioxidant effects in your body. But presence in the cup and activity in the body are separated by the entire obstacle course. That antioxidant has to survive a bath in hydrochloric acid (your stomach, pH 1.5 to 3.5). It has to slip through the fatty membrane of your intestinal wall. It has to run the gauntlet of your liver’s demolition enzymes before they chop it into metabolites. It has to reach the specific tissue where oxidative damage is occurring — your neurons, your hepatocytes, your pancreatic beta cells. And it has to arrive in sufficient concentration to matter. Each of these steps is a potential point of failure, and most health articles pretend none of them exist.
ADMET screening does not answer all of these questions. But it answers the first one — Are the molecular properties compatible with absorption? — and it gives us a framework for thinking about the rest.
For coffee’s fifteen key bioactives, the answer to that first question is encouraging. They all have molecular passports. They all pass the initial screening. This puts them in a fundamentally different category from, say, the large tannins in red wine, or the complex polysaccharides in mushrooms, or the massive polymeric pigments in many fruits and vegetables. Many plant compounds fail at the very first checkpoint. Coffee’s small-molecule bioactives do not. If Laura asked me today what I’d found, I would tell her: The obstacle course is real. And your coffee clears the first wall.
So here is where we stand.
The fifteen key bioactive compounds in your coffee — caffeine, the chlorogenic acids, the diterpenes cafestol and kahweol, trigonelline, and the constellation of phenolic acids — all have molecular properties that, in pharmaceutical terms, make them plausible oral bioactives. They are small enough, lipophilic enough, and possess the right hydrogen bonding characteristics to cross biological membranes. They pass the first filter.
This is a necessary foundation for everything we will discuss in the remaining chapters of this section. If these compounds failed Lipinski’s Rule — if they were too large, too polar, too sticky with water — then there would be little point in asking whether they reach the liver, the brain, or the heart. They would be biologically interesting compounds trapped in the gut, interacting with your microbiome (as melanoidins do) but not entering your systemic circulation.
But they pass. The door is open. And that single fact — that every major bioactive in your morning cup has the molecular credentials to enter your bloodstream — is what separates coffee from the parade of “superfoods” that make headlines and then quietly disappear. Turmeric’s curcumin? Brilliant in a test tube, nearly unabsorbable in your gut. Resveratrol from red wine? Metabolized to rubble before it reaches your blood. Many plant compounds are pharmacologically fascinating and biologically irrelevant. Coffee’s compounds are not. They get through.
The harder questions come next. Among these fifteen compounds, which ones can reach the brain? This turns out to depend on a second barrier — the blood-brain barrier — which is far more selective than the gut wall. Some coffee compounds are predicted to cross it. Others, despite passing every other checkpoint, are stopped cold.
That second barrier, and the molecular property that determines who passes and who does not, is the subject of the next chapter.
Next: Chapter 12 — “The Blood-Brain Barrier: Coffee’s Final Checkpoint”
Whether adding fat to coffee changes how you absorb its compounds
You'll Need
- Brewed black coffee
- Coconut oil or butter (1 tsp)
- Two mornings
- Notebook
Do This
- Day 1: Drink black coffee on an empty stomach. Note onset of alertness, peak, and duration.
- Day 2: Add 1 tsp coconut oil or butter to the same coffee, blend or stir vigorously. Drink on empty stomach. Note the same timeline.
- Compare your two curves — look for differences in onset speed and duration.
- Note any difference in how long you feel the effects.
What's Happening
Fat-soluble compounds in coffee (including some diterpenes and certain polyphenol metabolites) absorb more effectively when co-ingested with lipids. Fat slows gastric emptying, which can delay caffeine onset but extend its absorption window. This is the pharmacokinetic principle behind "bulletproof coffee" — though the effect varies significantly between individuals.
“The blood-brain barrier is the final frontier. What makes it through changes how you think, feel, and remember.”